Autour du documentaire Une étude qui dérange, Dr Gardenal, Pr Perronne, Dr Fouché et Del Bigtree viennent questionner le calendrier vaccinal infantile et ouvrir un questionnement plus large sur les enjeux de santé et l’affaiblissement de nos populations. A Lyon, dans le ventre de la baleine, la discussion invite à devenir chacun des porteurs du message. https://youtu.be/PKNzlCgOQa8?si=4vRH6G9jBixZ32qU An Inconvenient Truth ?
2026-06-25 – [CSI] Présentation
Dans cette conférence présentée au Conseil Scientifique Indépendant (CSI), le Dr Michel Cucchi, docteur en médecine, sociologue et master en biologie moléculaire et microbiologie, retrace l’histoire méconnue de la militarisation des sciences du vivant. De la naissance de la microbiologie moderne aux programmes contemporains de recherche biologique à risque, cette intervention explore les liens entre progrès scientifiques, secret d’État, expérimentations humaines et stratégies militaires.
À travers une analyse historique documentée, Michel Cucchi revient notamment sur :
▪️ l’émergence des armes biologiques au XXe siècle ;
▪️ les programmes japonais, allemands, américains, soviétiques et chinois ;
▪️ les expérimentations humaines menées au nom de la science ou de la guerre ;
▪️ la prolifération des arsenaux biologiques durant la Guerre froide ;
▪️ les enjeux actuels de transparence, de contrôle et de démilitarisation de la recherche. Une réflexion essentielle sur les dérives possibles de la recherche scientifique lorsqu’elle est intégrée à des logiques militaires, industrielles ou géopolitiques.
Avec : Michel Cucchi Louis Fouché Hélène Banoun
📅 Conférence du Conseil Scientifique Indépendant (CSI)
Chapitres:
Introduction
La naissance de la microbiologie moderne
Les premières armes biologiques
Expérimentations humaines et guerre biologique japonaise
L’Allemagne nazie : eugénisme et expérimentations
Les programmes américains et la Guerre froide
Le programme soviétique Biopreparat
La stratégie chinoise et la fusion militaro-civile
Contrôle des recherches à risque et perspectives
Questions et échanges
Mots-clés: armes biologiques, guerre biologique, microbiologie, militarisation du vivant, recherche à risque, biotechnologies, expérimentation humaine, Conseil Scientifique Indépendant, CSI, Michel Cucchi, Louis Fouché, Hélène Banoun, santé publique, bioéthique, histoire des sciences, guerre froide, armes de destruction massive, transparence scientifique
Hashtags #CSI #MichelCucchi #LouisFouché #HélèneBanoun #ArmesBiologiques #Microbiologie #Bioéthique #RechercheScientifique #SantéPublique #Histoire
Vous avez déjà vu un ciné débat en multiplex dans plus de 50 villes en même temps ?
Événement exceptionnel – Samedi 6 juin à 20h
« UNE ÉTUDE QUI DÉRANGE » : le film qui a fait trembler l’industrie pharmaceutique arrive en ciné-débat géant !
Sur tous nos canaux:
https://www.linkedin.com/feed/update/urn:li:ugcPost:7468687887354474496
https://www.facebook.com/events/967459636340362
https://www.facebook.com/events/1872273493452846
https://t.me/reinfocovid_officiel
https://crowdbunker.com/v/zsvAt138
Pour la première fois en France, Del Bigtree (producteur de Vaxxed, figure majeure de la liberté de choix aux États-Unis), Louis Fouché (anesthésiste-réanimateur, lanceur d’alerte) et Vincent Pavan (mathématicien et statisticien) se réunissent en direct pour un débat sans filtre après la projection.
Au programme :
Projection du film « Une étude qui dérange »
Débat en multiplex avec Del Bigtree (depuis les USA), Louis Fouché et Vincent Pavan à Paris
Questions du public en salle et dans les 50+ villes connectées
Un rendez-vous historique pour tous ceux qui refusent la censure, qui exigent la vérité sur les vaccins, la santé et les libertés fondamentales.
Paris : salle de cinéma en centre-ville (adresse exacte communiquée après inscription)
Partout en France : plus de cinquante collectifs participent en multiplex simultané.
Samedi 6 juin – 20h00
Ouvert à tous – Réservation obligatoire (places limitées)
Mots-clés SEO : ciné débat Paris, Une étude qui dérange, Del Bigtree Paris, Louis Fouché, Vincent Pavan, Vaxxed France, liberté vaccinale, débat vaccins, santé liberté, cinéma vérité, soirée débat Paris, événement santé juin 2026 2026
« Autisme et neuro-diversité: des pistes pour avancer »
Samedi 25 avril à Bruxelles & dimanche 26 avril à Paris

CONGRES
AUTISME DES PISTES POUR AVANCER
26 AVRIL PARIS
8h30-9h00 Ouverture des portes
9h- 9h10 Accueil et Ouverture :le mot du président de l’Association
9h10 – 9h30 Dr Louis Fouché : Tout seul on va plus vite, ensemble on va plus loin
9h30 Brian Hooker Autism an overview on the current research and science
10h15 Dr Eric Menat Quand des médecins bâtissent avec les familles les prises en soin – ça marche
10h45 – 11h00 pause
11h00 – 11h30 Dr Martine Gardénal Autisme, la fragilisation des terrains
11h30- 11h45 Sylvie Bennet Une approche pluridisciplinaire…rêvons un peu !
11h45- 12h30 Table ronde 1
Modération : Dr Louis Fouché
Dr Marie Grenet, Martine Gardénal, Sylvie Bennet, Brian Hooker + questions du public
12h30 – 14h00 Pause repas – networking – librairie
14h00 Mary Holland Autism advocacy and care: lessons learned and ways to go
14h45 -15h15 Senta Depuydt Agir sans attendre : réaliser la mise en commun de nos ressources et compétences.
15h15 – 15h30 Marie Perrazi-Touzet Plaidoyer pour un parcours de santé dès le diagnostic
15h30-15h45 Christine Buscailhon Comment lever les obstacles à la mise en place d’une alimentation adaptée ?
15h45 – 16h00 pause
16h00- 16h45 Table ronde 2
Mary Holland, Brian Hooker, Senta Depuydt, Marie Perrazi,-Touzet, Sylvie Bennet, Martine Gardénal, Christine Buscailhon + public
Modération : Louis Fouché
16h45 Louis Fouché : Autisme, quelles pistes pour avancer ? Synthèse et mise en mouvement.
17h30 Fin
Autisme, troubles du neurodéveloppement, TDA/H, troubles du langage : le
nombre des « diagnostics » et des « atypiques » connaît une hausse
spectaculaire en Europe et aux USA, et la France a fait de la santé mentale
une priorité pour les 5 prochaines années. Les enjeux sont immenses :
santé, éducation, inclusion, prévention, coût pour la société, mais aussi
liberté de soin et respect de la singularité de chaque personne.
Malheureusement, les parcours de soin restent souvent trop limités, en
particulier la prise en charge des problèmes de santé associés aux différents
diagnostics. Il reste difficile de mettre en place un soutien au niveau de
l’alimentation, de médecines complémentaires ou d’approches innovantes,
alors que des personnes restent en souffrance, sans l’écoute de leurs
besoins.
Les deux journées « Autisme : des pistes pour avancer » visent à rassembler
des parents, des médecins et des intervenants pour unir leurs compétences
et expériences dans l’accompagnement des personnes à besoins
spécifiques. Elle s’adressent à tous ceux qui souhaitent une approche plus
humaine et globale de la neuro-diversité : troubles de l’attention,
hypersensibilités, troubles anxieux, TDAH.
Le 25 avril à Bruxelles et le 26 avril à Paris, rejoignez-nous pour deux
journées d’espoir et d’action avec :
– Mary Holland (présidente de Children’s Health Defense) et Brian S. Hooker
(directeur scientifique, épidémiologiste). Science actuelle, politiques de
santé aux USA et ressources.
– Dr Louis Fouché (anesthésiste-réanimateur, parrain du congrès), voix libre
et humaniste de la médecine ;
– Dr Martine Gardénal : autisme, évolution des terrains
– Des parents et professionnels engagés : Senta Depuydt (compétence
parentale, ressources) Christine Buscailhon (microbiote & alimentation),
Marie Perrazi-Touzet (prise en charge de santé précoce ), Sylvie Bennet
(approche pluridisciplinaire) et Caroline Duchène (communication pour les
non parlants).
Au programme : des témoignages bouleversants, des données scientifiques,
approches complémentaires (intestin, nutrition, environnement, éducation
respectueuse) et des pistes pratiques pour avancer vraiment.
2 journées placées sous le signe de la rencontre, de l’échange et de la mise en
commun des savoirs, parce que chaque enfant et chaque adulte « différent »
mérite le maximum pour sa santé, son bien-être et son autonomie. Rendez-
vous le 25 et 26 Avril – Inscriptions ouvertes ! »
Un nouvel examen des études qui sous-tendent la limite imposée par la FDA à la quantité d’adjuvant en aluminium autorisée par dose de vaccin révèle que cette limite, fixée il y a plusieurs décennies, a été déterminée en fonction de la capacité de l’adjuvant à générer une réponse immunitaire, et non pas en fonction du risque que l’aluminium représente pour la santé.
17 septembre 2025
Au milieu des années 1900, les organismes de réglementation américains ont fixé la limite de l’adjuvant aluminium dans les vaccins à 0,85 milligramme (mg) par dose. Les organismes de réglementation n’ayant jamais modifié cette limite, les Centers for Disease Control and Prevention (CDC) affirment aujourd’hui que les adjuvants à base d’aluminium « sont utilisés en toute sécurité dans les vaccins depuis plus de 70 ans ».
Mais comment les autorités de réglementation ont-elles déterminé la limite de 0,85 mg par dose ? Et quelles preuves existe-t-il aujourd’hui qu’il est sûr d’exposer les enfants qui suivent le calendrier vaccinal recommandé par le CDC à des quantités d’aluminium allant jusqu’à cette limite ?
Ce n’est pas parce que des adjuvants en aluminium ont été utilisés dans les vaccins pendant des décennies qu’il s’ensuit logiquement que cette exposition à une neurotoxine connue est inoffensive – surtout si l’on considère qu’en plus des vaccins individuels, les enfants sont exposés à des quantités cumulées d’aluminium par le biais de plusieurs vaccins figurant dans le calendrier des CDC.
Pour étayer son affirmation, le CDC déclare que « les vaccins contenant des adjuvants font l’objet d’essais cliniques pour en vérifier l’innocuité et l’efficacité avant d’être autorisés à être utilisés aux États-Unis ».
Le CDC cite une page web de la Food and Drug Administration (FDA) des États-Unis, qui indique également que les vaccins contenant de l’aluminium « ont un profil de sécurité démontré au cours de nombreuses décennies d’utilisation ».
Selon la page web de la FDA, « lorsqu’elle évalue la sécurité et l’efficacité d’un vaccin, la FDA considère les adjuvants comme un composant du vaccin ; ils ne sont pas approuvés séparément ».
Le problème de ce raisonnement est que les essais cliniques des vaccins ne sont pas conçus pour détecter les effets néfastes à long terme – par exemple, le développement ultérieur d’allergies, de maladies auto-immunes ou de troubles du développement neurologique – de cette exposition à l’aluminium.
La déclaration de la FDA selon laquelle les adjuvants ne sont pas approuvés séparément pour leur sécurité revient à admettre qu’il existe une pénurie de données toxicologiques pour étayer l’affirmation selon laquelle l’injection aux enfants de vaccins contenant un adjuvant à base d’aluminium est inoffensive.
Un article publié le 26 août dans la revue Environmental Toxicology and Pharmacology met en évidence ce manque d’études de sécurité. L’article, intitulé « Les limites réglementaires de la teneur en aluminium des vaccins n’ont pas été fixées sur la base d’études toxicologiques« , est rédigé par les chercheurs français Loïc Angrand, Ph.D., Romain K. Gherardi et Guillemette Crépeaux, Ph.D.
Comme le révèlent les auteurs, la limite de 0,85 mg par dose, fixée il y a plusieurs décennies, était basée sur des considérations immunologiques – et non sur des données démontrant que cette quantité n’est pas toxique lorsqu’elle est injectée à des enfants. Cette limite n’a jamais été destinée à indiquer une quantité d’aluminium pouvant être considérée comme inoffensive pour les enfants.
Pourquoi les fabricants de vaccins utilisent-ils des adjuvants aluminium ?
Un adjuvant est une substance utilisée dans les vaccins pour stimuler une réponse immunitaire plus inflammatoire, ce qui entraîne une plus grande production d’anticorps en aval.
Les fabricants de vaccins utilisent depuis longtemps l’aluminium comme adjuvant privilégié pour répondre aux exigences réglementaires en matière d’immunogénicité, c’est-à-dire un certain niveau d’anticorps dans le sang considéré comme protégeant contre le virus (ou la bactérie) ciblé par le vaccin.
Dans le cadre réglementaire actuel, les titres d’anticorps servent généralement de mesure de substitution de l’immunité – même si aucun niveau d’anticorps n’a été scientifiquement corrélé à la protection contre l’infection ou la maladie.
Par exemple, le vaccin anticoquelucheux avec adjuvant en aluminium produit par GlaxoSmithKline Biologics (GSK) a été approuvé par la FDA sur la base de tests sanguins conçus pour mesurer la réponse en anticorps aux antigènes anticoquelucheux inclus dans le vaccin.
GSK admet dans la pièce jointe à l’emballage du vaccin qu’il ne s’agit pas d’une approche scientifiquement validée. La notice précise :
« Le rôle des différents composants produits par B. pertussis dans la pathogenèse de la coqueluche ou dans l’immunité contre la coqueluche n’est pas bien compris. »
Ces informations sont légalement requises dans les notices d’emballage car, bien que les fabricants aient bénéficié d’une large immunité juridique en vertu du National Childhood Vaccine Injury Act de 1986 – qui a transféré la charge financière des dommages dus aux vaccins de l’industrie pharmaceutique à l’État (c’est-à-dire aux consommateurs contribuables) – ils peuvent toujours être poursuivis en justice pour avoir fait des déclarations frauduleuses concernant la sécurité et l’efficacité de leurs produits.
Comment les régulateurs ont-ils déterminé la limite de 0,85 mg/dose ?
Comme le soulignent Angrand et ses collègues dans leur article, pendant de nombreuses décennies, les adjuvants à base d’aluminium « ont été considérés comme sûrs par les organismes de réglementation » malgré l’accumulation de preuves d’effets toxiques chez l’animal et l’homme.
Ces preuves de nocivité comprennent des observations cohérentes selon lesquelles les adjuvants à base d’aluminium peuvent induire des maladies auto-immunes et que l’aluminium est biopersistant et transloque du site d’injection vers d’autres tissus et organes – y compris le cerveau, où il s’accumule.
En 2019, le réseau Informed Consent Action Network (ICAN) a envoyé des demandes d’information (FOIA) aux National Institutes of Health (NIH) et à l’Agency for Toxic Substances and Disease Registry (ATSDR), une sous-agence du CDC, pour demander les études que les agences ont utilisées « pour établir la sécurité de l’injection de nourrissons et d’enfants » avec des adjuvants à base d’aluminium.
Les deux agences ont répondu à la demande d’accès à l’information en déclarant qu’aucun document de ce type n’avait pu être localisé.
Selon l’examen du dossier documentaire effectué par les chercheurs, la teneur maximale de 0,85 mg d’aluminium a été fixée par le NIH en 1968. L’autorité réglementaire sur les produits biologiques, qui comprend les vaccins, a été transférée en 1972 à la FDA, qui a conservé la limite de 0,85 mg.
En creusant davantage, les chercheurs ont obtenu, par le biais d’une requête FOIA, deux documents du NIH qui ont servi de base à l’établissement de cette limite supérieure.
Le premier document, datant de 1947, concernait la fabrication de l’anatoxine diphtérique. Le second, datant de 1952, concerne la fabrication de l’anatoxine tétanique.
Ces documents révèlent que les autorités réglementaires ont fixé la limite de 0,85 mg en se fondant non pas sur des données toxicologiques, mais sur des données d’immunogénicité, c’est-à-dire sur la quantité d’adjuvant en aluminium nécessaire pour induire la réponse immunitaire voulue.
Le document de 1947 stipule que « dans tous les cas, la quantité d’aluminium utilisée doit être la plus faible possible pour atteindre l’objectif visé ».
Une déclaration similaire figure dans le document de 1952. Aucun de ces documents ne traite de la toxicité de l’aluminium.
Les seules références en matière de sécurité étaient les exigences relatives à l’essai de l’anatoxine respective sur des souris et des cobayes, au moins deux animaux de chaque espèce, avec un suivi d’au moins une semaine pour détecter les symptômes immédiatement évidents ou la mort, ainsi qu’un test de « désintoxication » impliquant « au moins quatre animaux » observés pendant un mois.
Les chercheurs ont conclu qu’il « est difficile de voir comment les textes [de ces documents] pourraient servir de base fiable pour garantir la sécurité » des vaccins avec adjuvant en aluminium recommandés systématiquement pour les enfants aujourd’hui par le CDC.
La limite réglementaire « établie il y a plus de 60 ans », résument les auteurs, était « initialement basée sur l’efficacité immunologique des seules anatoxines diphtérique et tétanique, plutôt que sur des évaluations toxicologiques solides » des adjuvants à base d’aluminium.
La FDA admet que la sécurité n’est pas une priorité
Le NIH et la FDA ont également reconnu explicitement la note des chercheurs français.
Lors d’un atelier sur l’aluminium dans les vaccins en 2000, le Dr Michael Gerber du NIH a demandé au Dr Norman Baylor de la FDA s’il pouvait expliquer comment les régulateurs étaient parvenus à la limite de 0,85 mg par dose.
Baylor a répondu : « Malheureusement, je ne peux pas l’expliquer ».
Il a ajouté qu’ils avaient « essayé de le découvrir » à partir des « archives historiques », mais « sans succès ».
Deux ans plus tard, Baylor et ses collègues ont publié un article dans la revue Vaccine dans lequel ils admettent que la limite de 0,85 mg par dose « a été choisie de manière empirique à partir de données qui ont démontré que cette quantité d’aluminium renforçait l’antigénicité et l’efficacité du vaccin ».
En d’autres termes, elle était fondée sur la nécessité perçue de générer un niveau élevé d’anticorps, et non sur des considérations relatives à la sécurité de l’injection d’une neurotoxine connue à des enfants.
« L’innocuité des adjuvants à base d’aluminium a été prouvée », ont néanmoins conclu Baylor et ses collègues, en se basant sur le raisonnement circulaire selon lequel « les vaccins utilisant des adjuvants à base d’aluminium ont un profil d’innocuité démontré depuis plus de six décennies ».
Selon les chercheurs français, « en l’état, la limite de 0,85 mg d’Al [aluminium] par dose de vaccin semble justifiée par un précédent historique et non par une investigation scientifique rigoureuse correspondant aux calendriers de vaccination actuels ».
En d’autres termes, le règlement établissant une limite de 0,85 mg par dose n’a jamais eu pour but d’indiquer une quantité d’aluminium pouvant être considérée comme inoffensive pour les enfants.
Elle n’est pas non plus totalement arbitraire.
Il s’agissait simplement d’un plafond qui garantissait une quantité d’aluminium suffisante pour provoquer la réponse immunitaire souhaitée sans être immédiatement réactogène :il s’agissait d’éviter que les signes de toxicité aiguë se manifestent presque immédiatement dans les modèles animaux.
Les scientifiques et les médias appliquent deux poids deux mesures lorsqu’ils interprètent des données d’observation
Dans leur article, Angrand et ses coauteurs soulèvent des inquiétudes croissantes quant à l’évolution de la situation :
- Augmentation de l’exposition à l’aluminium « chez les nouveau-nés, les nourrissons, les enfants, les adolescents et les femmes enceintes ».
- Utilisation d’injections contenant de l’aluminium à la place de placebos dans les essais cliniques.
- Variabilité de la teneur en aluminium mesurée dans les flacons de vaccins n’ayant aucun rapport avec les quantités déclarées par les fabricants.
- Absence de divulgation de la nature chimique d’adjuvants spécifiques.
- L’absence d’études épidémiologiques de grande envergure permettant d’examiner les effets à long terme sur la santé.
Parmi les études épidémiologiques qui ont été menées, on peut citer une étude du CDC publiée dans Academic Pediatrics en 2023. L’étude « a révélé une association significative entre l’exposition cumulative à l’Al associée aux vaccins avant l’âge de 24 mois et l’incidence de l’asthme persistant à l’âge de 24 à 59 mois. »
La publication de cette étude a été accompagnée des assurances habituelles selon lesquelles il ne s’agit que de données d’observation, d’une étude « imparfaite » à laquelle nous ne devrions pas accorder trop d’importance en raison de ses limites méthodologiques.
Cela contraste avec la nature des rapports sur les études d’observation qui n’établissent aucun lien entre les vaccins et les effets néfastes. Ces études sont régulièrement qualifiées de méthodologiquement si solides que nous pouvons considérer leurs résultats comme absolument concluants.
Une récente étude danoise déclarant l’aluminium sûr s’attire des critiques
C’est le cas d’une récente étude menée au Danemark par Niklas Worm Andersson, M.D., Ph.D., un chercheur danois du Statens Serum Institut à Copenhague.
L’étude a examiné les effets des vaccins contenant l’adjuvant aluminium et un grand nombre de problèmes de santé chez l’enfant, notamment les maladies atopiques et allergiques, l’auto-immunité et les troubles du développement neurologique.
Les médias ont salué l’étude comme une preuve concluante que l’aluminium contenu dans les vaccins est inoffensif pour les enfants.
Mais comme le notent Angrand et ses collègues, « les débats méthodologiques en cours sur l’évaluation de l’exposition, les critères d’exclusion, les ajustements utilisés et la cohérence des données d’une correction ultérieure justifient pour l’instant une interprétation prudente ».
Les commentaires des scientifiques sur l’étude danoise, publiée le 15 juillet dans Annals of Internal Medicine, comprennent un large éventail de critiques :
- L’absence d’une cohorte de contrôle non exposé
- Exclusion effective des enfants à haut risque.
- Résultats biologiquement peu plausibles d’un effet protecteur de l’aluminium.
- Irreproductibilité de leurs résultats en raison de la non-divulgation des données et de la modélisation.
- L’absence de contrôle de la tendance des enfants présentant un risque plus élevé de subir les conséquences à recevoir moins de vaccins (parfois appelé « biais des vaccinés sains« ).
- Une durée de suivi inférieure à l’âge moyen du diagnostic pour de nombreux résultats.
- Une contradiction entre l’affirmation des auteurs selon laquelle ils n’ont trouvé aucune preuve de nocivité et les associations statistiquement significatives montrées dans une version corrigée de leur supplément.
L’étude danoise est biaisée, selon certains critiques
De nombreuses analyses critiques de l’étude ont été rédigées. Dans une critique publiée le même jour que l’étude, le chercheur et auteur James Lyons-Weiler, Ph.D., a souligné comment les chercheurs danois ont « ajusté » leurs données pour le nombre de visites chez le médecin généraliste avant l’âge de 2 ans, traitant ainsi à tort ce nombre comme s’il s’agissait d’une variable indépendante.
Cela introduit un biais de collision, qui se produit lorsque deux variables influencent toutes deux une troisième – la « collision ». Le fait de traiter un facteur de collision comme un facteur de confusion et de l’ajuster peut dissimuler des associations réelles entre les expositions et les dommages.
Dans ce cas, les vaccinations peuvent entraîner une augmentation des visites chez le médecin en raison des effets indésirables. Les symptômes précoces des effets néfastes des vaccins peuvent également entraîner une augmentation des visites chez le médecin. Le nombre de visites est donc influencé à la fois par l’exposition et par le résultat.
En outre, le nombre de visites peut à son tour influencer ces deux facteurs : plus de visites peuvent entraîner plus de vaccinations et plus de diagnostics, créant ainsi une boucle de rétroaction qui se renforce d’elle-même.
Comme il ne s’agit pas d’une variable indépendante, le fait d' »ajuster » leurs données en fonction du nombre de visites de cette manière a biaisé l’étude en faveur de l’absence de lien.
Ceci, ainsi que d’autres décisions méthodologiques, pourrait expliquer pourquoi l’étude a trouvé des associations négatives statistiquement significatives malgré l’invraisemblance biologique de l’exposition à l’aluminium comme protecteur contre les maladies atopiques et allergiques, l’auto-immunité et les troubles du développement neurologique.
RFK Jr. demande la rétractation d’une étude danoise
Dans un article publié le 30 juillet par le Brownstone Institute, le mathématicien tchèque Tomas Fürst, Ph.D., et la chercheuse médicale danoise Vibeke Manniche, M.D., Ph.D., ont souligné, parmi d’autres problèmes de l’étude, la manière dont les enfants présentant un risque plus élevé de résultats mesurés ont été effectivement exclus.
Ils ont également noté que le supplément original a été remplacé par une version corrigée montrant « une association statistiquement significative entre certains troubles du développement neurologique – en particulier l’autisme et le TDAH – et l’exposition à l’aluminium des vaccins ».
Le secrétaire américain à la santé et aux services sociaux (HHS), Robert F. Kennedy Jr., a demandé la rétractation de l’étude dans un article publié par Trial Site News le 1er août, dans lequel il résumait les nombreuses façons dont l’étude était biaisée en faveur de l’hypothèse nulle.
Alors que les auteurs de l’étude danoise affirment qu’ils « n’ont pas trouvé de preuves » d’un risque accru, Kennedy a également noté que leurs données supplémentaires corrigées montrent des associations significatives. Comme il l’a écrit :
« Les données montrent une augmentation statistiquement significative de 67 % du risque de syndrome d’Asperger par augmentation de 1 mg de l’exposition à l’aluminium chez les enfants nés entre 2007 et 2018.
« Par rapport au groupe d’exposition modérée, pour 10 000 enfants de la cohorte d’exposition à l’aluminium la plus élevée, il y a eu 9,7 cas de plus de troubles neurodéveloppementaux, 4,5 cas de plus de troubles autistiques et 8,7 cas de plus de la catégorie plus large des troubles du spectre autistique. »
C’est ce que corrobore une critique de l’étude par des scientifiques de Children’s Health Defense (CHD) publiée le 5 août sur Preprints.org, un serveur en ligne pour les articles qui n’ont pas encore fait l’objet d’un examen par les pairs.
Dans cet article, Karl Jablonowski, Ph.D., chercheur principal de CHD, et Brian S. Hooker, Ph.D., responsable scientifique de CHD, soulignent comment, dans le matériel complémentaire original de l’étude, « les diagnostics de développement neurologique des enfants les plus malades ont été supprimés », alors que « dans le matériel complémentaire mis à jour, où ces diagnostics ont été inclus » – et après avoir rétabli les enfants omis – les données « ont révélé une association entre l’exposition à l’aluminium et les maladies dévastatrices du développement neurologique ».
Qualifiant l’étude de « travail défectueux avec de bonnes preuves de fraude », Jablonowski et Hooker ont fait écho à l’appel de Kennedy en faveur de la rétractation de l’étude.
Des chercheurs danois refusent de rétracter leur étude
Les rédacteurs de la revue ont toutefois refusé de rétracter l’étude, affirmant dans un commentaire public le 11 août que l’étude « n’a trouvé aucune preuve d’une association claire » et qu’elle est exempte de toute « erreur sérieuse » susceptible d’invalider leurs conclusions ou d’indiquer une faute scientifique.
Dans un article reprenant les nombreuses failles qui, ensemble, indiquent que l’étude a été conçue pour ne pas établir de lien, Lyons-Weiler décrit la revue comme étant impliquée dans le « toilettage du discours plutôt que dans l’arbitrage scientifique neutre ».
Dans un commentaire public sur leur étude répondant aux critiques, Andersson et ses collègues ont fait valoir que les associations significatives montrées dans leurs données supplémentaires pouvaient être rejetées parce que les enfants nés avant 2002, qui ont reçu moins de vaccins contenant de l’aluminium, ne pouvaient pas appartenir au groupe le plus exposé, et que le fait de réexécuter l’analyse sans ces enfants effaçait la signification statistique.
Cependant, leur analyse principale a également comparé les expositions et les résultats entre les cohortes de naissance, y compris celles nées avant 2002. L’étude a été conçue pour vérifier si les enfants plus exposés à l’aluminium, nécessairement dominés par les cohortes nées plus tard, présentaient des risques plus élevés de subir les conséquences de l’exposition à l’aluminium.
Le CDC s’est appuyé sur une analyse obsolète et discréditée de la sécurité des vaccins avec adjuvant en aluminium.
Pour étayer leur conclusion selon laquelle les vaccins avec adjuvant aluminium sont inoffensifs, les chercheurs danois ont cité sans esprit critique une étude de la FDA réalisée par Robert J. Mitkus, Ph.D., et al. sur laquelle le CDC s’appuie pour étayer son affirmation selon laquelle « la quantité d’exposition à l’aluminium chez les personnes qui suivent le calendrier vaccinal recommandé est faible et n’est pas facilement absorbée par l’organisme ».
Le CDC accompagne cette affirmation du raisonnement circulaire habituel selon lequel les adjuvants à base d’aluminium « sont utilisés en toute sécurité dans les vaccins depuis des décennies ».
Dans leur article révélant l’absence de données de sécurité sous-jacentes à la limite réglementaire de 0,85 mg par dose, Angrand et ses collègues font allusion à cette étude de la FDA en déclarant :
« En outre, les points de vue théoriques dépassés et mal documentés qui soutenaient ces affirmations ont été remis en question par la compréhension croissante de la nature des nanoparticules, de leur persistance imprévue à long terme dans les cellules immunitaires, de leur capacité à se propager à partir du site d’injection dans tout le corps, y compris le cerveau, de leur neurotoxicité dans les modèles animaux, et par les préoccupations croissantes concernant leur rôle potentiel dans les troubles du développement neurologique et les troubles allergiques ».
La référence fournie est « Analyse critique des études de référence sur la toxicocinétique des adjuvants à base d’aluminium« , par Jean-Daniel Masson, Ph.D., et al, et publiée dans le Journal of Inorganic Biochemistry en 2018.
Les auteurs de cette critique, dont les associés d’Angrand, Crépeaux et Gherardi, ont noté que Mitkus et ses co-auteurs ont adopté de manière inappropriée un « niveau de risque minimal » (MRL) défini pour la prise orale quotidienne d’aluminium soluble chez les souris – et non pour la forme particulaire insoluble de l’aluminium injecté aux enfants.
Les chercheurs de la FDA ont également effectué des « calculs erronés de la durée d’absorption » et ont ignoré la diffusion systémique des particules d’aluminium, y compris dans le cerveau, où elles peuvent provoquer une neuroinflammation.
Lorsque j’ai demandé à Mme Crépeaux quel était le problème de citer l’étude de Mitkus de la FDA comme preuve de l’innocuité des adjuvants à base d’aluminium, elle m’a répondu :
« Le principal problème est que des études plus récentes évaluées par des pairs ont expliqué pourquoi Mitkus se trompe. Le CDC devrait fonder ses recommandations sur une littérature actualisée, et Mitkus se trompe à plusieurs égards, comme nous l’avons expliqué en 2018 – il y a déjà sept ans ! »
Dans leur nouvel article examinant les origines de la limite de 0,85 mg par dose, Crépeaux et ses collègues concluent qu’il existe un « besoin urgent d’études pharmacocinétiques et toxicologiques indépendantes » en rapport avec le calendrier de vaccination actuel du CDC.
Ignorer les échecs alarmants de la recherche existante pour explorer et encore moins démontrer la sécurité, remarquent les auteurs, « ne les ferait pas disparaître ».
Les points de vue et les opinions exprimés dans cet article sont ceux des auteurs et ne reflètent pas nécessairement ceux de Children’s Health Defense.
Jeremy R. Hammond est un chercheur et un journaliste indépendant qui s’attache à dénoncer la propagande mensongère du courant dominant qui sert à faire accepter des politiques gouvernementales néfastes.
Le virus varicelle-zona (appelé VZV, pour varicella-zoster virus) est un herpès-virus persistant, également appelé HHV-3 (human herpes virus 3) ou herpès virus humain type 3, responsable chez l’être humain de la varicelle et du zona (herpes zoster).
Un virus persistant est un virus qui reste dans l’organisme à long terme, parfois à vie, sans forcément provoquer de symptômes constants. Il peut :
- Rester latent (inactif) puis se réactiver.
- Se multiplier à bas bruit, échappant au système immunitaire.
Le VZV ne doit pas être confondu avec d’autres herpès qui appartiennent cependant à la même famille comme
- Herpès labial dû à herpes simplex virus 1 ou HSV1. Il est responsable du bouton de fièvre. Sa réactivation peut être très fréquente, parfois plusieurs fois par an.
- L’herpès Génital dû à herpes simplex virus 2 ou HSV2
Le zona et la varicelle sont donc provoqués par le même virus, le virus varicelle-zona (VZV).
La varicelle représente la primo-infection, souvent contractée pendant l’enfance et considérée comme une maladie infantile bénigne. La varicelle se caractérise par une éruption cutanée avec des démangeaisons, de la fièvre et de la fatigue. La plupart des personnes contractent la varicelle pendant l’enfance et développent une immunité naturelle efficace et protectrice.
Le zona est une réactivation du virus VZV, survenant plus tard dans la vie car le VZV peut rester dans les ganglions rachidiens sous forme inactive malgré la guérison de la varicelle. Cette réactivation intervient souvent chez les personnes dont le système immunitaire est affaibli mais d’autres facteurs plus ou moins clairs interviennent. La réactivation du VZV se fait depuis un ganglion rachidien et provoque une éruption cutanée localisée au dermatome innervé par le ganglion rachidien. L’éruption est donc unilatérale, c’est-à-dire sur un seul côté du corps. L’éruption cutanée peut être très douloureuse et c’est le symptôme le plus gênant mis en avant par les patients qui cherchent donc à s’en protéger.
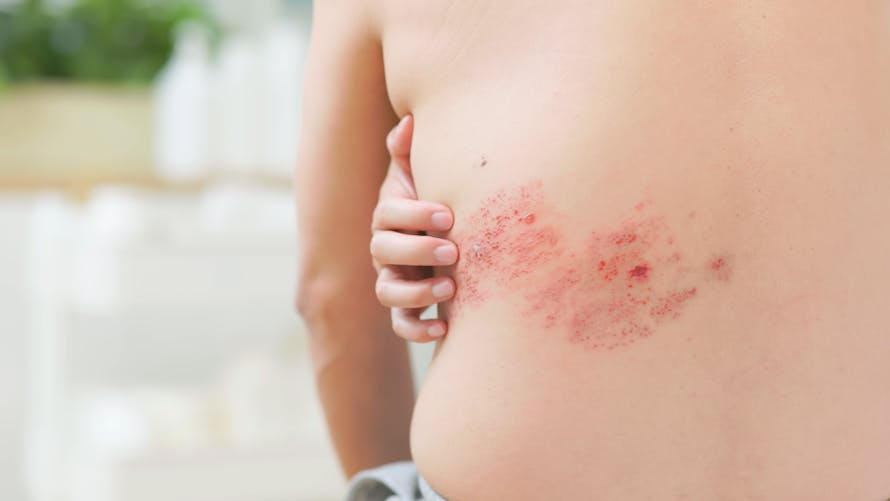
https://www.santemagazine.fr/sante/fiche-maladie/zona-177347and feel pain
Le vaccin Shingrix
Shingrix est un vaccin recombinant de la glycoprotéine E avec adjuvant (AS01B), destiné à la prévention du zona et des névralgies post-zostériennes chez les adultes de 50 ans et plus, ainsi que chez les adultes de 18 ans et plus présentant un risque accru de zona. Il est administré en deux doses, espacées de deux mois. Depuis le 14 décembre 2024, il est remboursé à 65% par l’Assurance Maladie pour les personnes concernées par les recommandations de la HAS [4]:
- chez les adultes de 65 ans et plus
- chez les adultes de 18 ans et plus présentant une immunodépression particulière
La demande de vaccination contre le zona est suffisamment importante pour que le vaccin Shingrix connaisse en 2025 des tensions d’approvisionnement. Il est contingenté en ville. [0]
Mais que disent les essais cliniques ?
Essai clinique du vaccin Shingrix
Un essai clinique a été publié en 2016 sous le titre :« Efficacité du vaccin Shingrix contre l’herpès-zona chez les adultes âgés de 70 ans ou plus » [1]
Les auteurs expliquent que la vaccination est une option intéressante pour réduire la charge de morbidité due à l’herpès-zona et à ses complications chez les personnes âgées.
Les auteurs (dont des employés du fabricant GSK) s’expliquent pourtant mal les raisons théoriques de l’efficacité de ce vaccin. Comment expliquer qu’en injectant un antigène de ce même virus et donc en restimulant le système immunitaire on puisse empêcher la réactivation du virus latent ?
Leur thèse est que l’inoculation d’un antigène viral (la glycoprotéine E contenue dans le vaccin) va restimuler l’immunité cellulaire, en particulier les lymphocytes T CD4+.
Les cellules T, en particulier les lymphocytes T CD4+ et CD8+, seraient essentielles pour maintenir cette latence en surveillant et en supprimant toute tentative de réactivation du virus. Ces lymphocytes T8 reconnaissent les antigènes viraux présentés par les cellules infectées et limiteraient la réplication virale. Cette explication de l’éventuelle efficacité du vaccin n’est pas pourtant prouvée.
Comment a été évaluée l’efficacité ?
Avant de commencer, il faut préciser que l’étude n’a pas intégré de patients ayant déjà eu un épisode de Zona. De même, les personnes immunodéprimées, qui sont parmi les plus susceptibles de subir des Zonas, ont aussi été exclues.
Ce choix est discutable car ces groupes semblent les plus légitimes à demander une vaccination efficace contre le Zona.
Il s’agit d’évaluer la protection contre les cas de zona.
Un cas suspect de zona est défini comme une éruption cutanée unilatérale accompagnée de douleur à l’exclusion de tout diagnostic alternatif. Si un cas n’est pas suspecté d’être un zona par l’investigateur, son évolution n’est pas suivie et il disparaît des statistiques (p46 protocole). Chaque cas suspect est identifié comme cas de zona si la PCR est positive ou bien si les cliniciens du comité de certification des cas votent en majorité pour un cas confirmé.
L’efficacité est aussi mesurée contre les douleurs post-zostériennes définies comme des douleurs sévères apparaissant ou s’aggravant plus de 90 jours après une éruption de zona.
Malheureusement de nombreuses modifications du protocole concernant les tests PCR et la définition des cas ne peuvent exclure que certains cas positifs chez les vaccinés et négatifs chez les placebo aient peut-être été requalifiés au cours de l’essai. [8]
Mais il y a plus grave. Le vaccin et le placebo n’avaient pas le même aspect. Malgré ce que les chercheurs affirment dans l’étude :
« The investigators, participants, and persons responsible for evaluating the study end points were unaware of whether HZ/su or placebo had been administered. »
« Les chercheurs, les participants et les personnes chargées d’évaluer les critères d’évaluation de l’étude ne savaient pas si HZ/su ou un placebo avait été administré.”
La personne qui injectait et le participant étaient donc au courant du groupe auquel le sujet était affecté (vacciné ou placebo). C’est une étude « observer-blinded ». Et le protocole a aussi été modifié sur ce point au cours de l’essai.
On peut donc avoir des doutes sur l’évaluation des « cas » de zona des participants à l’essai. Le biais est évident puisque les patients savent s’ils ont reçu le vaccin ou pas.
Chaque sujet est suivi pendant au moins 30 mois ; l’étude est terminée lorsque le nombre de cas de zona et de douleurs post-zostériennes prévus est atteint et lorsque 75% des sujets ont été suivis pendant au moins 36 mois.
L’incidence du zona est de 10 à 12,5 cas pour 1000/an chez les plus de 70 ans [9], compatible avec l’incidence de 9,2 dans le groupe placebo.
L’efficacité annoncée contre le zona est de 87,7% en réduction du risque relatif et de 2,39% en réduction du risque absolu. Elle est calculée sur une durée de 4 ans après la vaccination. On ne sait pas ce qui se passe après !
Il faut maintenant examiner en détails les effets indésirables rapportés par les auteurs.
Sécurité du vaccin
1 décès est attribué au vaccin dans l’essai clinique.
Il y a eu 29 décès dans le groupe placebo contre 17 dans le groupe vacciné (les 2 groupes sont de la même taille). Parmi des 17 décès vaccinés, 13 sont des morts subites (donc non expliquées et représentent 76% des décès enregistrés). Seules 4 morts subites sont retrouvées chez les placebo (14%).
La question se pose immédiatement de comprendre pourquoi dans 76% des décès du groupe vacciné, les médecins n’ont pas été capables de déterminer la cause de la mort alors que cela ne représente que 14% dans le groupe placebo.
Le déséquilibre des décès laisse penser que le groupe placebo était plus fragile que le groupe vacciné, Les morts subites excédentaires dans le groupe vacciné posent question, même si ce n’est sans doute pas statistiquement significatif.
Autre déséquilibre marquant, celui des cancers du poumons. Le déséquilibre en défaveur du vaccin pose également question : 18 cancers du poumon chez les vaccinés contre 10 chez les placebo (tableau S7). Mais les résultats sont incohérents car ne correspondent pas au tableau S5 ou on trouve 22 vaccinés et 13 placebo.
Parmi les effets indésirables graves, on note d’autres déséquilibres :
- 26 sepsis (infection grave) chez les vaccinés et 17 chez les placebo.
- Infection à staphylocoque: 4 vaccinés et 1 placebo.
- Les ostéoarthrites : 26 sont signalées chez les vaccinés contre 17 chez les placebo, est-ce à rapprocher des fractures par compression de la colonne vertébrale (6 vaccinés et 1 placebo) ?
Tous ces déséquilibres posent beaucoup de questions.
Finalement, la liste des effets indésirables graves reconnus en lien avec le vaccin :
- 1 infarctus du myocarde
- 1 colite
- 1 pancréatite
- 1 granulomatose éosinophilique avec polyangéite
- 4 infections (dont 1 otite herpétique)
- 1 leucémie aigüe
- 1 Guillain-Barré
- 1 eczéma
- 4 désordres généraux
- 1 lymphadénite
- 1 colite
- 1 pancréatite
Selon le rapport de 2018 de l’EMA [10], dans les essais cliniques, un décès a été attribué au vaccin.
Dans le même rapport, un autre effet indésirable grave est signalé : il s’agit d’une maladie auto-immune, le purpura thrombocytopénique (le suivi des maladies auto-immunes a été ajouté par un amendement au protocole de l’essai en 2014 ; l’essai se terminant en 2014).
L’EMA n’a donc pas tenu compte des autres effets indésirables graves reconnus en lien avec le vaccin dans l’essai de 2016. Les raisons de ces choix discutables ne sont pas connus.
D’autres effets indésirables non détectés dans les essais cliniques ont été recensés après la commercialisation du vaccin. Le Shingrix est capable, par exemple, de provoquer également un lupus. [2]
Autres problèmes concernant le Shingrix
D’autres problèmes s’ajoutent, notamment au niveau de la composition du vaccin. Le vaccin est préparé sur des cellules d’ovaires de hamster chinois et peut donc contenir des fragments d’ADN de hamster. De plus, il est adjuvanté avec l’AS01B, connu pour sa toxicité. Et pour finir, il contient du PEG 80 qui peut faciliter l’entrée dans les cellules des fragments d’ADN résiduels.
Conclusion
Quel est l’intérêt de vacciner contre une maladie que l’on a déjà contractée alors que l’immunité naturelle acquise par la maladie est définitive comme le confirme Santé Publique France [5] ?
En effet, plus de 90% des adolescents et 98% des femmes enceintes sont immunisés naturellement contre le virus de la varicelle zona en France [6]
En France, il est pourtant recommandé de vacciner uniquement les adolescents ou jeunes adultes qui n’ont pas eu la varicelle dans l’enfance.[7]
Et contrairement à ce qu’affirme le Figaro [3], le vaccin n’est pas destiné aux personnes immunodéprimées (voir l’avis de la HAS [4])
En conclusion, pourquoi les autorités françaises recommandent-elles, contradictoirement, de ne vacciner que les adolescents qui n’auraient pas eu la varicelle dans l’enfance et de vacciner tous les plus de 65 ans (qui l’ont certainement eue)?
Ce vaccin n’est pas sans danger et la démonstration de son efficacité peut être mise en doute!
[0] https://lequotidiendupharmacien.fr/exercice-pro/shingrix-victime-de-son-succes
[1] https://pubmed.ncbi.nlm.nih.gov/27626517/
[2] https://pubmed.ncbi.nlm.nih.gov/40500571/
[6] https://vaccination-info-service.fr/Les-maladies-et-leurs-vaccins/Varicelle
[7] https://vaccination-info-service.fr/Les-maladies-et-leurs-vaccins/Varicelle
[8] https://www.nejm.org/doi/suppl/10.1056/NEJMoa1603800/suppl_file/nejmoa1603800_protocol.pdf
[9] https://www.mesvaccins.net/web/news/4961-la-vaccination-contre-le-zona-recommandee-de-65-a-79-ans)
[10] https://www.ema.europa.eu/en/documents/overview/shingrix-epar-medicine-overview_pl.pdf
Depuis 2021, le grand public connait la technologie ARNm pour les vaccins. Cette technologie qui n’avait jamais été utilisée, y compris à petite échelle, a été propulsée sur l’ensemble de la population française et mondiale.
L’efficacité et la sécurité de cette technologie pose maintenant des questions évidentes. Avant même de répondre aux questions soulevées par l’utilisation des ARNm, une nouvelle technologie, un dérivé, apparait : saARNm ou self-amplified ARNm.
Pourtant, ces « nouveaux nouveaux » vaccins sont déjà à l’étude sur l’homme comme avec cette publication
Self-amplified ARNm ou saARNm
Pour le décrire rapidement, l’ARNm auto-amplifié (saARNm) est une version, selon ses concepteurs, « améliorée » de la technologie de l’ARN messager (ARNm) utilisée dans les vaccins Pfizer et Moderna contre le COVID que tout le monde connait depuis 2021.
La différence est cependant majeure et renforce le questionnement déjà évoqué pour les vaccins ARNm :
- Quantité de protéine produite.
- Durée de vie de l’ARNm.
Les saARNm contiennent, comme pour la technologie ARNm, l’information génétique pour provoquer la synthèse d’une protéine (comme la protéine Spike du SARS-CoV-2). Cette protéine, en théorie, est censée provoquer une immunisation protectrice.
La différence repose sur la propriété du saARNm de se recopier (répliquer) lui-même (self amplified) à l’intérieur des cellules grâce à l’ajout de l’information nécessaire à cette amplification dans le saARNm. Ainsi, à partir d’une molécule d’ARNm, plusieurs sont formées et chacune induira la production de plusieurs protéines cibles.

Adapté de https://pubmed.ncbi.nlm.nih.gov/33093657/
Les auteurs précisent que l’utilisation de ce type de technologie, qui amplifie l’ARNm dans les cellules de l’hôte permet de réduire « les coûts de production »
De fait, la quantité d’ARNm finale dans les cellules est inconnue. Par conséquent, la quantité de protéine produite l’est aussi.
La quantité de produit actif est pourtant un paramètre essentiel d’un produit pharmaceutique.
Un pharmacien ne peut pas délivrer un produit sans connaitre sa composition.
Article R.4235-61
« Le pharmacien ne peut dispenser un médicament que s’il a pu s’assurer de sa provenance, de sa composition, de sa posologie et de ses conditions de conservation. »
En conséquence ce produit, ce vaccin, ne devrait pas être délivré. Mais ici, cela ne pose de problème à personne.
Remarquons que, pour les vaccins ARNm de Pfizer et Moderna, c’était la même problématique puisque la quantité de produit actif, la protéine SPIKE, reçue par le patient était inconnue. La quantité d’ARNm était, elle aussi, inconnue à cause de procédés de fabrications imparfaits. Le produit n’aurait jamais dû être délivré par les pharmaciens.
Les auteurs sont très clairs
Les auteurs de la publication que nous vous proposons d’étudier rapportent l’étude d’un vaccin saRNA contre le SARS-CoV-2 en Ouganda. Les auteurs sont fiers de préciser qu’il s’agit d’un des premiers essais de ce type en Afrique. L’étude souligne également la faisabilité de mener des essais avancés de vaccins en Afrique comme si cela n’était pas une évidence sauf à penser que les Africains sont moins compétents que nous… [1]
« Les résultats soutiennent la sécurité et l’immunogénicité des vaccins saRNA et leur potentiel en tant que solution évolutive pour la COVID-19 et d’autres maladies infectieuses »
Il n’y a aucun doute sur leur enthousiasme.
- Sécurité
- Immunogénicité.
Immunogénicité
Dans cette étude de phase 1, il a été choisi de ne suivre que le taux d’anticorps induits par la vaccination. Ni les infections, ni la maladie n’ont été retenues comme marqueurs.
Les résultats montrent effectivement une immunogénicité (production d’anticorps spécifiques) pour les patients séronégatifs.
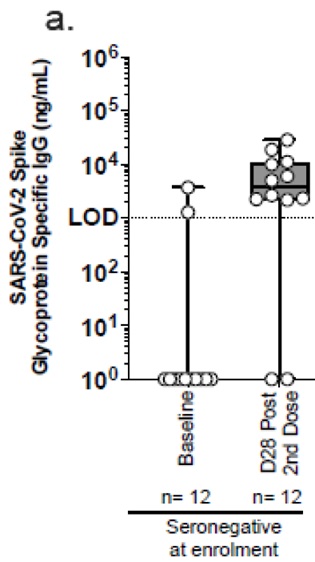
En revanche, il n’y a pas d’effet notable pour les patients précédemment déjà infectés (pas d’augmentation du taux d’anticorps). [2]
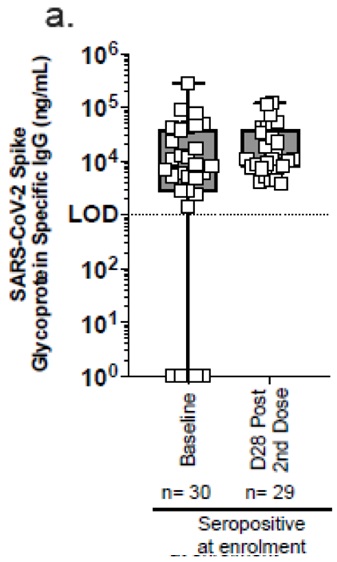
Mais le plus important n’est pas là.
La HAS confirme que le taux (titre/concentration) n’est pas corrélé à la protection contre le COVID. D’ailleurs les soignants avec des anticorps naturels après infection se sont vu refuser une réintégration à cause de ce motif.

[3]
Dans un second temps, les auteurs ont mesuré l’efficacité des anticorps par un test de neutralisation du pseudovirus 2 semaines après la seconde vaccination. Ce test n’est, lui non plus, pas pertinent puisque cette méthode n’a jamais été validée, jamais reconnue comme fiable pour permettre d’évaluer l’efficacité d’un vaccin.
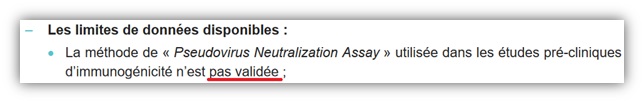
[3]
En résumé, les auteurs ont prouvé qu’un antigène viral induisait une réponse immunitaire humorale.
Rien de plus.
Rien d’intéressant en 2025.
Mais aucune efficacité sur la mortalité, les formes graves n’été mise en évidence puisqu’elle n’a même pas été recherchée.
Sécurité
Les auteurs indiquent que l’essai a démontré que le vaccin était bien toléré, sans événements indésirables graves signalés.
Regardons ces résultats.
Les résultats des effets indésirables considérés comme graves sont présentés dans le tableau suivant qui regroupe les 42 participants dont 30 initialement étaient séropositifs pour le SARS-CoV-2 donc déjà infectés au moins 1 fois.
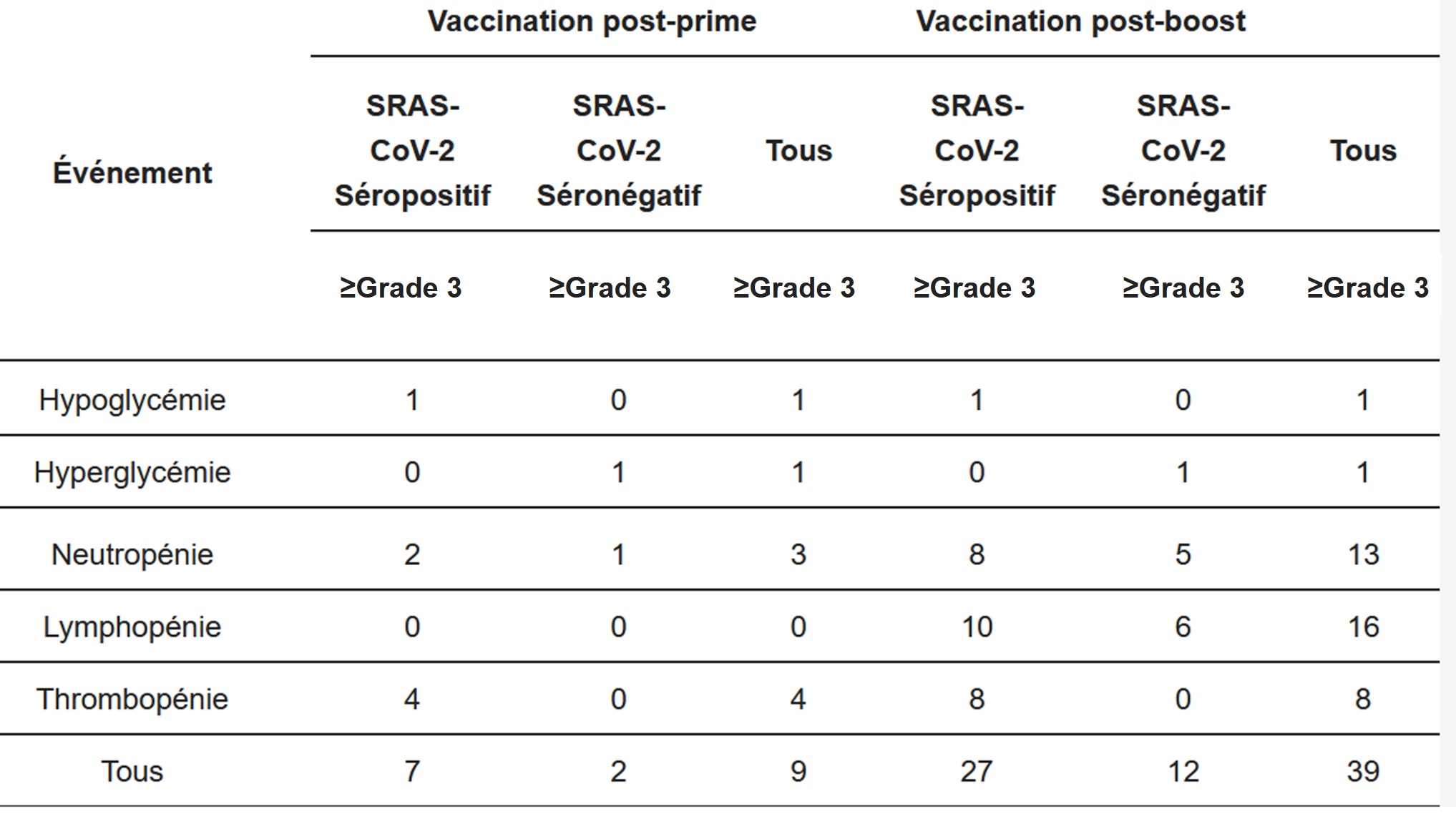
Les patients déjà infectés (séropositifs) sont 3,5 fois plus touchés par les effets indésirables graves au moment de la première dose et 2,25 fois plus au moment de la 2eme dose.
Les auteurs ne semblent pas interloqués par ce constat qui montre pourtant un risque bien plus important lors de la vaccination de patients déjà immunisés par infection naturelle.
Concernant maintenant le nombre d’effets indésirables graves, les chiffres sont alarmants.
Au moment de la première dose, les auteurs rapportent 9 évènements pour 42 patients. Cela représente un taux de 21% si on considère que chaque patient n’a qu’un effet indésirable.
Au moment de la deuxième dose, les auteurs rapportent 39 évènements pour 42 patients. Cela représente un taux de 93% si on considère que chaque patient n’a qu’un effet indésirable (39/42 = 93%).
Les auteurs ne précisent malheureusement pas la distribution des effets indésirables graves sur les patients. Il n’est donc pas exclu qu’un patient présente plusieurs effets indésirables graves en même temps. Dans ce cas, en reprenant les calculs, on obtient un taux minimum de 38% d’effets indésirables graves au niveau de la 2eme dose. En effet, on peut affirmer qu’il y a eu au moins 16 personnes individuelles atteintes, puisque les cas de lymphopénie sont forcément sur 16 patients différents, soit 16/42 = 38%.
Ainsi, il y a entre 38% et 93% d’effets indésirables graves. Pourtant les auteurs assurent que tout va bien !
Qui peut considérer qu’il n’y a pas de problème ?
De plus, les auteurs ne semblent pas gênés par la nature des symptômes principaux essentiellement :
- Neutropénie
- Lymphopénie
- Thrombopénie
Mais encore mieux, les auteurs concluent :
« Aucun des effets intermédiaires cliniques de grade 3 ou supérieur ou des anomalies biologiques n’a été attribué au vaccin. »
Circulez, il n’y a rien à voir !
Qui peut le comprendre ?
Conclusion
L’analyse des résultats de l’étude ne permet pas de comprendre l’enthousiasme des auteurs. Surtout au niveau de la sécurité avec des taux supérieurs à 38%, au minimum 4 personnes sur 10 injectées
Mais sachez tout de même que :
- Au Japon, un vaccin à ARN messager auto-amplifié contre la Covid a été approuvé pour la première fois en novembre 2023 avant d’être utilisé dans une campagne de vaccination en octobre 2024.
- En Europe, ce même vaccin a été approuvé par la Commission européenne en février 2025. [4]
- Le vaccin, contre la grippe aviaire, ARNm auto-amplifiant est utilisé depuis septembre/octobre 2023 en France pour les canards. Son autorisation a été renouvelée pour 2 ans jusqu’au 25 Mars 2027.
Evidemment aucun test n’a été effectué sur la capacité de l’ARNm auto amplifié injecté aux animaux à être transmis au consommateur.
Mais cela ne gène pas ceux qui piquent.
[1] https://doi.org/10.3390/vaccines13060553
Sujet : Alerte sanitaire sur les ARNm anti-Covid
Invité : Dr Louis FOUCHÉ – Médecin Anhestésiste-Réanimateur, Master II éthique et Anthropologie de la Santé
Discutante : Hélène BANOUN – Pharmacienne et ancienne Chargé de recherches INSERM
Sujet : Contrats vaccins de Ursula Von Der LEYEN
Invité : Maître Arnaud DURAND – Avocat au Barreau de Paris depuis plus de dix ans, spécialisé en matière de libertés individuelles, droits fondamentaux, santé et environnement.
Discutant : Hélène BANOUN – Pharmacienne et ancienne Chargé de recherches INSERM
Animateur : Laurent MUCCHIELLI – Sociologue Directeur de recherche au CNRS
Sujet : Contrats vaccins de Ursula Von Der LEYEN
Invité : Maître Arnaud DURAND – Avocat au Barreau de Paris depuis plus de dix ans, spécialisé en matière de libertés individuelles, droits fondamentaux, santé et environnement.
Discutante : Hélène BANOUN – Pharmacienne et ancienne Chargé de recherches INSERM
Animateur : Laurent MUCCHIELLI – Sociologue Directeur de recherche au CNRS
![]()
Partenaires
![]()
![]()
![]()
Suivez-nous sur les réseaux !
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()