Autour du documentaire Une étude qui dérange, Dr Gardenal, Pr Perronne, Dr Fouché et Del Bigtree viennent questionner le calendrier vaccinal infantile et ouvrir un questionnement plus large sur les enjeux de santé et l’affaiblissement de nos populations. A Lyon, dans le ventre de la baleine, la discussion invite à devenir chacun des porteurs du message. https://youtu.be/PKNzlCgOQa8?si=4vRH6G9jBixZ32qU An Inconvenient Truth ?
2026-06-25 – [CSI] Présentation
Dans cette conférence présentée au Conseil Scientifique Indépendant (CSI), le Dr Michel Cucchi, docteur en médecine, sociologue et master en biologie moléculaire et microbiologie, retrace l’histoire méconnue de la militarisation des sciences du vivant. De la naissance de la microbiologie moderne aux programmes contemporains de recherche biologique à risque, cette intervention explore les liens entre progrès scientifiques, secret d’État, expérimentations humaines et stratégies militaires.
À travers une analyse historique documentée, Michel Cucchi revient notamment sur :
▪️ l’émergence des armes biologiques au XXe siècle ;
▪️ les programmes japonais, allemands, américains, soviétiques et chinois ;
▪️ les expérimentations humaines menées au nom de la science ou de la guerre ;
▪️ la prolifération des arsenaux biologiques durant la Guerre froide ;
▪️ les enjeux actuels de transparence, de contrôle et de démilitarisation de la recherche. Une réflexion essentielle sur les dérives possibles de la recherche scientifique lorsqu’elle est intégrée à des logiques militaires, industrielles ou géopolitiques.
Avec : Michel Cucchi Louis Fouché Hélène Banoun
📅 Conférence du Conseil Scientifique Indépendant (CSI)
Chapitres:
Introduction
La naissance de la microbiologie moderne
Les premières armes biologiques
Expérimentations humaines et guerre biologique japonaise
L’Allemagne nazie : eugénisme et expérimentations
Les programmes américains et la Guerre froide
Le programme soviétique Biopreparat
La stratégie chinoise et la fusion militaro-civile
Contrôle des recherches à risque et perspectives
Questions et échanges
Mots-clés: armes biologiques, guerre biologique, microbiologie, militarisation du vivant, recherche à risque, biotechnologies, expérimentation humaine, Conseil Scientifique Indépendant, CSI, Michel Cucchi, Louis Fouché, Hélène Banoun, santé publique, bioéthique, histoire des sciences, guerre froide, armes de destruction massive, transparence scientifique
Hashtags #CSI #MichelCucchi #LouisFouché #HélèneBanoun #ArmesBiologiques #Microbiologie #Bioéthique #RechercheScientifique #SantéPublique #Histoire
Des affirmations plus que contestables
L’article affirme que « la vitamine A a suscité une hausse des intoxications signalées chez les enfants » et que la cause en serait « une interview télévisée du 4 mars 2025, où le secrétaire à la Santé Robert F. Kennedy Jr. vante l’huile de foie de morue et la vitamine A » pour le traitement de la rougeole.
Des confusions majeures entretenues par le journaliste
Le rapport du centre antipoison américain rapporte une hausse des « expositions » et non des intoxications !
Entre le 1er janvier et le 31 mars 2025, 86 expositions pédiatriques à la vitamine A ont été signalées aux centres antipoison américains, soit une augmentation de 38,7 % par rapport à la même période de l’année précédente.[1]
Cependant, le rapport précise également qu’aucune augmentation de la gravité des cas n’a été observée et qu’aucun effet majeur n’a été signalé. Les auteurs rappellent en outre que toutes les expositions ne conduisent pas à une intoxication ou à une maladie, et que certains signalements peuvent concerner des produits cosmétiques contenant de la vitamine A, comme le rétinol.
En résumé, pour la période janvier-mars 2025 :
- 86 cas d’exposition à la vitamine A ont été signalés ;
- 0 cas d’effet majeur ont été rapportés.
Présenter ces chiffres comme une vague d’intoxications est donc au minimum discutable.
Aucune augmentation de la gravité des expositions n’a été observée !
Aucun effet majeur n’a été signalé
La vitamine A est un traitement recommandé pour éviter les formes graves de la rougeole.
L’article omet également un élément important : la vitamine A n’est pas présentée par les autorités sanitaires comme une substance dangereuse à éviter absolument.
Au contraire, l’administration de vitamine A fait partie des recommandations médicales pour les patients atteints de rougeole, notamment chez les enfants hospitalisés, afin de réduire le risque de complications graves. L’OMS le confirme [3] :
« Tous les enfants ou adultes atteints de rougeole devraient recevoir deux doses de suppléments de vitamine A, administrées à 24 heures d’intervalle »
Les données citées par les centres antipoison sont donc également compatibles avec l’hypothèse que davantage d’enfants ont reçu une supplémentation en vitamine A dans le contexte d’une augmentation des cas de rougeole (incidence USA 2024 : 0,00008%, Incidence USA 2025 : 0,0007%).
Sans que cela n’entraîne d’effets toxiques graves.
Des chiffres à replacer dans leur contexte
L’article ne fournit aucun élément de comparaison permettant d’apprécier l’importance réelle des 86 expositions signalées.
À titre de comparaison, les centres antipoison américains ont recensé :
- 52 855 expositions au paracétamol en 2023 ;
- 51 769 expositions au paracétamol en 2024 ;
- 38 615 expositions au paracétamol au cours des premiers mois de 2025.
Le paracétamol est par ailleurs l’une des principales causes d’intoxications médicamenteuses graves et de décès par surdosage aux États-Unis.
Or personne ne considère pour autant que les recommandations médicales visant à utiliser le paracétamol pour faire baisser la fièvre devraient être interdites ou censurées.
Corrélation = causalité ?
L’article établit également un lien entre :
- l’intervention médiatique de Robert F. Kennedy Jr. ;
- l’augmentation des recherches Internet concernant la vitamine A ;
- l’augmentation des expositions déclarées à la vitamine A.
Or cette relation demeure purement une corrélation temporelle ou co-incidence.
De nombreux journalistes rappellent régulièrement, qu’une corrélation temporelle ne suffit pas à démontrer un lien causal, notamment lorsqu’il s’agit d’effets indésirables déclarés après une vaccination.
Il est donc surprenant de voir ce même principe méthodologique totalement oublié lorsqu’il s’agit d’établir un lien entre une intervention médiatique et l’augmentation d’intoxications imaginaires liés à la vitamine A.
Deux poids et deux mesures chez nos chers journalistes ?
Environ 1500 effets indésirables déclarés après le vaccin ROR
L’article omet également de dire que chaque année plusieurs centaines d’effets indésirables suite au vaccin ROR sont déclarés dans le VAERS ; soit bien plus que les cas d’exposition à la vitamine A.
Comme pour les déclarations aux centres antipoison, ces signalements ne démontrent pas à eux seuls un lien causal avec le produit concerné. Ils constituent des signaux nécessitant une évaluation et une investigation complémentaires.
En particulier en 2025, il y aurait eu pour le vaccin ROR, selon Vaccine Watch, 1629 effets indésirables déclarés dont 3 mentionnant un décès, 60 une hospitalisation, 173 un passage aux urgences et 26 un handicap [2].
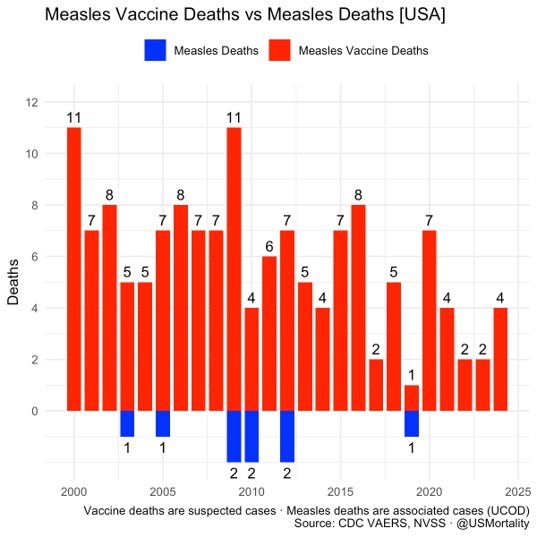
Il est donc incohérent de présenter les expositions déclarées à la vitamine A comme une preuve implicite de danger tout en rappelant, dans d’autres contextes, que les déclarations d’effets indésirables vaccinaux ne permettent pas à elles seules d’établir une causalité.
POINTS CLES
- Aucune augmentation des intoxications à la vitamine A selon les centres antipoison américains
- Seulement une augmentation des expositions sans aucune augmentation de la gravité des cas
- La vitamine A fait partie des traitements recommandés pour réduire le risque de complications graves chez les patients atteints de rougeole
Afin que chacun puisse se faire sa propre opinion, nous reproduisons ci-dessous la traduction automatique du communiqué officiel des centres antipoison américains.
Traduction du communiqué officiel des centres antipoison américain :
Les centres antipoison constatent une augmentation des expositions à la vitamine A chez les enfants pendant l’épidémie de rougeole
7 avril 2025 – 14 h 40
Shauna Devitt (Administratrice)
Ce qu’il faut savoir
La rougeole est une infection virale très contagieuse qui provoque actuellement des flambées épidémiques aux États-Unis. Des informations récentes suggèrent que certaines personnes utilisent la vitamine A ou l’huile de foie de morue pour prévenir l’infection par la rougeole. La meilleure façon de prévenir la rougeole reste la vaccination par le vaccin ROR (rougeole, oreillons, rubéole).
Chez les patients atteints de rougeole, en particulier les enfants hospitalisés, une supplémentation en vitamine A est recommandée afin de réduire le risque de complications graves. Toutefois, avant d’administrer de la vitamine A ou tout autre complément alimentaire ou produit à base de plantes, il est conseillé de consulter un professionnel de santé.
Les Centres antipoison américains surveillent les cas de toxicité à la vitamine A signalés dans le National Poison Data System (NPDS). Entre le 1er janvier et le 31 mars 2025, 86 expositions pédiatriques à la vitamine A ont été rapportées aux centres antipoison américains, soit une augmentation de 38,7 % par rapport à la même période en 2024.
Cependant, malgré cette hausse du nombre de cas, aucune augmentation de la gravité des intoxications n’a été observée, et aucun effet grave n’a été signalé en 2025. Toutes les expositions ne conduisent pas nécessairement à une maladie ou à une intoxication. De plus, les expositions à la vitamine A recensées dans le NPDS peuvent également provenir de produits cosmétiques contenant de la vitamine A, comme le rétinol.
Risques liés à une mauvaise utilisation de la vitamine A
Bien que la vitamine A soit généralement sûre lorsqu’elle est utilisée correctement, une consommation excessive peut entraîner une intoxication.
Les symptômes peuvent inclure :
- Nausées, vomissements, douleurs abdominales
- Maux de tête liés à une augmentation de la pression intracrânienne
- Douleurs osseuses
- Troubles de la vision
- Atteintes hépatiques (dommages au foie)
Points essentiels à retenir
La meilleure protection contre la rougeole est la vaccination par le vaccin ROR.
La vitamine A joue un rôle important dans le traitement de la rougeole, mais elle doit être utilisée avec prudence et sous surveillance médicale.
Une consommation excessive de vitamine A peut provoquer une toxicité grave.
En cas de suspicion de surdosage ou d’inquiétude concernant une exposition à la vitamine A, contactez un centre antipoison ou un professionnel de santé.
[1] Poison Centers Observe Increased Vitamin A Exposures in Children During Measles Outbreak ; Shauna Devitt • April 07, 2025 2:40 PM (https://poisoncenters.org/news-alerts/13484508)
[2] https://www.vaccinewatch.org/vaccines/mmr/2025
[3] https://www.who.int/fr/news-room/fact-sheets/detail/measles
Vous avez déjà vu un ciné débat en multiplex dans plus de 50 villes en même temps ?
Événement exceptionnel – Samedi 6 juin à 20h
« UNE ÉTUDE QUI DÉRANGE » : le film qui a fait trembler l’industrie pharmaceutique arrive en ciné-débat géant !
Sur tous nos canaux:
https://www.linkedin.com/feed/update/urn:li:ugcPost:7468687887354474496
https://www.facebook.com/events/967459636340362
https://www.facebook.com/events/1872273493452846
https://t.me/reinfocovid_officiel
https://crowdbunker.com/v/zsvAt138
Pour la première fois en France, Del Bigtree (producteur de Vaxxed, figure majeure de la liberté de choix aux États-Unis), Louis Fouché (anesthésiste-réanimateur, lanceur d’alerte) et Vincent Pavan (mathématicien et statisticien) se réunissent en direct pour un débat sans filtre après la projection.
Au programme :
Projection du film « Une étude qui dérange »
Débat en multiplex avec Del Bigtree (depuis les USA), Louis Fouché et Vincent Pavan à Paris
Questions du public en salle et dans les 50+ villes connectées
Un rendez-vous historique pour tous ceux qui refusent la censure, qui exigent la vérité sur les vaccins, la santé et les libertés fondamentales.
Paris : salle de cinéma en centre-ville (adresse exacte communiquée après inscription)
Partout en France : plus de cinquante collectifs participent en multiplex simultané.
Samedi 6 juin – 20h00
Ouvert à tous – Réservation obligatoire (places limitées)
Mots-clés SEO : ciné débat Paris, Une étude qui dérange, Del Bigtree Paris, Louis Fouché, Vincent Pavan, Vaxxed France, liberté vaccinale, débat vaccins, santé liberté, cinéma vérité, soirée débat Paris, événement santé juin 2026 2026
Dans cet entretien explosif, l’avocat suisse Philippe Kruse décortique les controverses juridiques, politiques et éthiques entourant la gestion mondiale de la crise de la COVID-19.
👉 Les sujets abordés incluent : les poursuites engagées contre Swissmedic ; l’autorisation d’urgence des vaccins contre la COVID ; l’influence croissante de l’Organisation mondiale de la Santé ; la censure, les traités relatifs aux pandémies et les pouvoirs d’urgence ; le lobbying pharmaceutique et les conflits d’intérêts ; la responsabilité démocratique et les politiques de santé publique. S’adressant à Louis Fouché, Philippe Kruse affirme que la pandémie a accéléré une dangereuse concentration de pouvoir au sein des institutions internationales. Il appelle à un audit international indépendant de la gestion de la COVID-19, du processus d’autorisation des vaccins et du rôle joué par les gouvernements et les agences sanitaires mondiales.
📄 Plainte officielle de Philippe Kruse contre Swissmedic : Plainte Corona – Site officiel https://plaintecorona.ch/ #OMS #COVID19 #PhilippeKruse #LouisFouche #TraitéPandémie #Vaccins #Swissmedic #SantéPublique #Liberté #Démocratie #ARNm #TraitéOMS #PolitiqueDeSanté #IndustriePharmaceutique #Pandémie #SantéMondiale #LibertéMédicale #EnquêteCOVID #DroitInternational #Censure
Dans cet épisode du **Conseil Scientifique Indépendant (CSI)**, le Dr **Louis Fouché** reçoit le philosophe **Reza Moghaddassi**, aux côtés de **Vincent Pavan** et **Laurent Mucchielli**, pour une réflexion approfondie sur la **philosophie des sciences**, la **médecine moderne** et les dérives du **scientisme**.
À travers une analyse claire et accessible, cette conférence explore les grandes transformations de notre rapport à la connaissance et à la vérité :
➡️ Différence entre **science et scientisme**
➡️ Limites de la biomédecine face à la **complexité du vivant**
➡️ Place des médecines complémentaires et de la **médecine intégrative**
➡️ Crise de confiance dans les institutions scientifiques
➡️ Enjeux de la **corruption systémique** dans la recherche
➡️ Importance du doute, de l’incertitude et du pluralisme
Dans un monde marqué par la crise sanitaire, cette discussion propose un regard critique sur la production du savoir scientifique et invite à repenser la médecine dans une approche plus **humaine, globale et ouverte**.
—
🎥 **Au programme :**
* Philosophie des sciences et épistémologie
* Critique du scientisme
* Médecine moderne vs approches complémentaires
* Complexité du vivant
* Science, pouvoir et intérêts économiques
* Vers une médecine plus intégrative
—
🌐 **Site officiel du Conseil Scientifique Indépendant :**
[https://conseil-scientifique-independant.org/](https://conseil-scientifique-independant.org/)
—
🔔 Abonnez-vous pour suivre les prochains épisodes du CSI
👍 Likez et partagez pour soutenir une information libre et indépendante
—
## 🔎 Mots-clés SEO (tags YouTube)
conseil scientifique indépendant, CSI louis fouché, reza moghadassi, vincent pavan, laurent mucchielli, philosophie des sciences, scientisme définition, médecine moderne critique, médecine intégrative, épistémologie science, crise covid analyse, corruption scientifique, industrie pharmaceutique critique, esprit critique science, vérité scientifique débat, paradigme scientifique, post modernité science, santé globale, médecine holistique, débat scientifique france, sciences et société, pensée critique, conférence santé, biologie moderne critique, système de santé crise, médecine et philosophie, savoir scientifique limites, fake news science

Un vaccin a pour objectif de diminuer les effets d’une infection. L’effet le plus significatif reste évidemment sur la mortalité. Le vaccin doit diminuer la mortalité de la maladie.
Personne ne contestera cette position.
Début 2021, il a été répété que les vaccins COVID étaient sûrs et efficaces. Les chiffres d’efficacité annoncés par les ministres, les journalistes, les médecins de plateaux télé étaient d’au moins 95%, parfois 100%. Y compris contre le variant DELTA ou OMICRON en 2022.
A partir de ces annonces, le calcul était facile. Il était facile de prévoir la mortalité COVID de 2021 en connaissant celle de 2020 en intégrant l’efficacité annoncée du vaccin.
Ce n’est évidemment pas aussi facile que ça car il y a des paramètres externes aux vaccins à prendre en compte.
- Premièrement, la population de 2021 n’est plus naïve vis-à-vis du COVID. Une immunité est donc déjà présente dans la population et doit diminuer naturellement la mortalité l’année suivante.
- Deuxièmement, les individus les plus fragiles de la population ont succombé à l’apparition de cette nouvelle infection en 2020. Le nombre de décès devrait donc être mécaniquement moins grand en 2021.
- Troisièmement, le virus s’adapte à son hôte et s’atténue. [1]
Une partie de la baisse de la mortalité COVID ne serait donc pas imputable seulement au vaccin mais à ces paramètres externes. Dans un premier temps, nous n’en tiendrons pas compte.
Les données de mortalité COVID pour la république Tchèque sont disponibles sur 5 ans et par classe d’âge. [2]
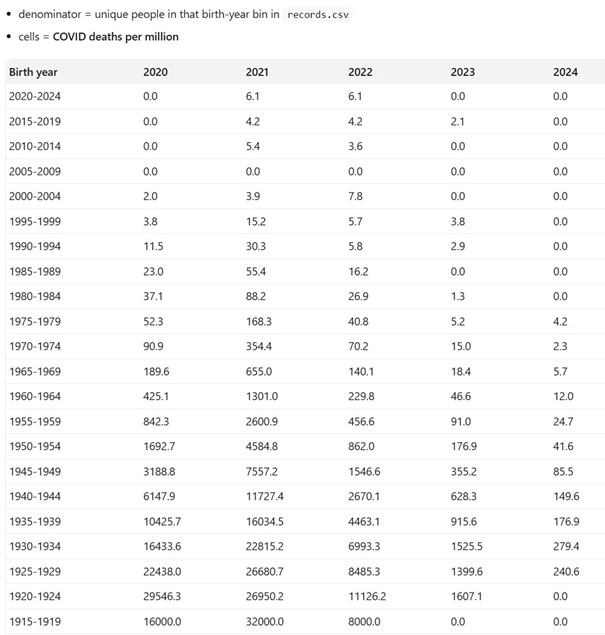
Prenons l’exemple des gens âgés de 65 à 70 ans en 2020 en République Tchèque. La mortalité COVID est mesurée à 1692 par million.
Ainsi, avec un vaccin efficace à 95%, la mortalité attendue sera donc de 85 décès par million.
Malheureusement, la mortalité mesurée en 2021 était de 4584 par million. La mortalité a été augmentée presque d’un facteur 3 (2,71 pour être précis).
Pour les moins de 25 ans
Pour les plus jeunes, la mortalité COVID était nulle en 2020 et pourtant présente en 2021.
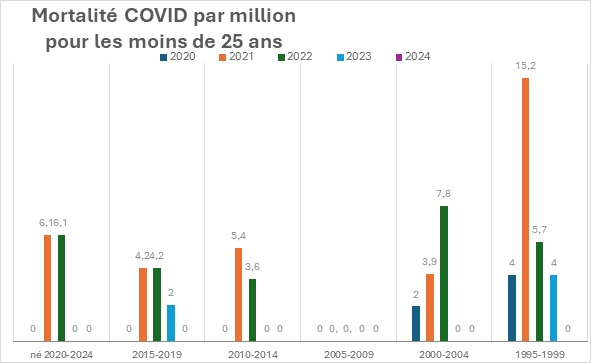
Pour les individus nés entre 2000 et 2004, qui ont été vaccinés plus tardivement, fin 2021, on remarque que l’augmentation de la mortalité COVID est encore plus importante en 2022. Elle est augmentée quasiment d’un facteur 4. Il y a eu 4 fois plus de décès en 2022 qu’en 2020 malgré la vaccination.
En résumé, sur ces classes d’âge, il n’y a pas eu de réduction de la mortalité mais une augmentation.
Une forte augmentation.
Parfois d’un facteur 4.
Pour les 26 à 55 ans
Pour les jeunes, entre 26 et 55 ans, l’augmentation de mortalité en 2021, par rapport à 2020 est comprise entre un facteur 2,6 à 3,9. Ces valeurs sont identiques aux classes d’âge précédentes.
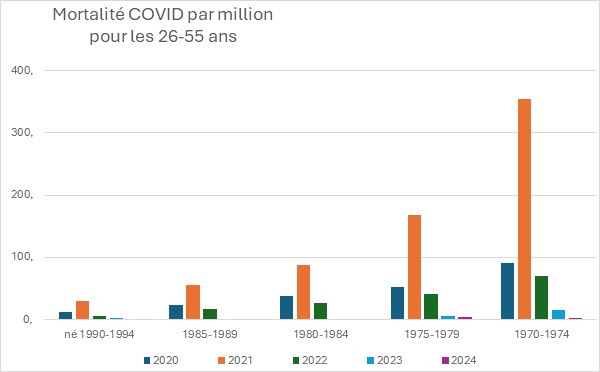
Il faut remarquer que plus l’âge est important, plus l’augmentation de la mortalité est importante.
Cette donnée est à mettre en parallèle du fait que la couverture vaccinale était aussi plus importante avec l’âge puisque les plus âgés se sont plus fait vacciner.
En résumé, il semble que plus la couverture vaccinale est importante, plus la mortalité a augmenté en 2021 (année avec vaccin) par rapport à 2020 (année sans vaccin).
Dans cette classe d’âge, la couverture vaccinale est d’environ 70%.
Mettre un graphique des couvertures vaccinales?
Pour les 55 à 85 ans
Pour les 55-85 ans, l’augmentation de la mortalité en 2021, par rapport à 2020 est comprise entre un facteur 1,5 à 3,5.
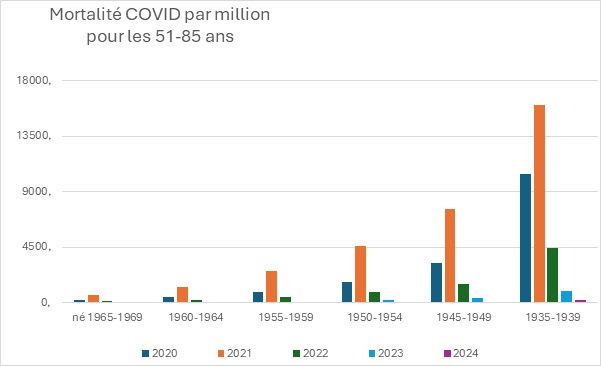
Le nombre de décès attribué au COVID a donc massivement augmenté en 2021, donc en période vaccinale, plus qu’en 2020 sans vaccin. La couverture vaccinale est pourtant d’environ 80% dans cette classe d’âge.
Pour les plus de 85 ans.
Le constat est le même chez les plus âgés : augmentation de la mortalité en période vaccinale comparé à la période de 2020 sans vaccin.
Il faut remarquer que pour les centenaires, il n’y a pas d’augmentation mais une baisse très modeste, et loin de 59%, de 10% de mortalité en 2021 par rapport à 2020. C’est la seule classe d’âge qui montre une réduction. En 2020, c’est la classe d’âge qui montre aussi la plus forte mortalité, c’est la classe d’âge la plus touchée. L’effet « moisson » de 2020 pourrait expliquer que la mortalité de 2021 ne la dépasse pas.
Bilan
Dans toutes les classe d’âge, sauf marginalement pour les centenaires, l’année 2021 a été marquée par une mortalité COVID bien plus importante qu’en 2020.
Dans toutes les classes d’âge, sauf pour les centenaires, l’utilisation de la vaccination COVID a été accompagnée par une mortalité COVID bien plus importante qu’en 2020.
Parfois d’un facteur 4.
Rappelez-vous du début : « Le vaccin doit diminuer la mortalité de la maladie. »
L’analyse de ces données de la République Tchèque ne permet assurément pas de voir une efficacité de la campagne massive de vaccination. La baisse de 95% attendue de la mortalité, annoncée par les médias, grâce à la vaccination n’a pas eu lieu.
Et de très loin
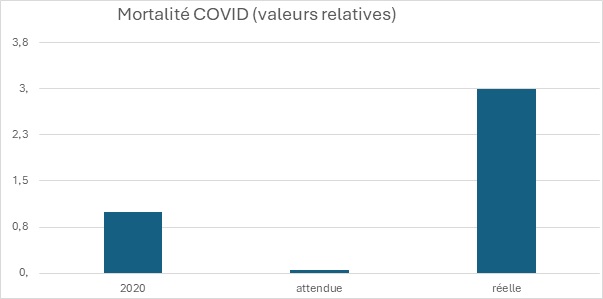
Mortalité COVID observée en 2020 (population totale), mortalité COVID attendue en 2021 en tenant compte de l’efficacité des vaccins Covid. Mortalité COVID réelle en 2021.
Revenons aux paramètres externes exposés précédemment. Leurs impacts attendus sur la baisse de mortalité ne sont même pas visibles. Ils sont engloutis dans l’augmentation de la mortalité COVID de 2021 par rapport à 2020.
Conclusion
La baisse de la mortalité naturelle (population non naïve, plus âgés déjà partis) attendue n’est pas visible.
La baisse de la mortalité attendue par la vaccination de masse n’est pas visible.
Mais pire encore, la mortalité, en plus de ne pas avoir baissé, a augmenté. Et parfois de 300% pour certaines classes d’âge.
Il est évident que ces données de la République Tchèque ne montrent pas d’effet protecteur de la vaccination. Il n’y a pas de baisse.
Il reste alors à expliquer d’où vient cette augmentation, parfois de 300%, des décès COVID à partir de l’apparition des vaccins COVID.
Les hypothèses sont les bienvenues…
Bibliographie
[1] (Banoun H. Evolution of SARS-CoV-2: Review of Mutations, Role of the Host Immune System. Nephron. 2021;145(4):392-403. doi: 10.1159/000515417. Epub 2021 Apr 28. PMID: 33910211; PMCID: PMC8247830. https://pubmed.ncbi.nlm.nih.gov/33910211/
[2] https://x.com/stkirsch/status/2050694612088709251/photo/1
An Inconvenient Study
Ciné-débats avec Del Bigtree, Clémence Houdiakova, Louis Fouché, Christian Perronne… et bien d’autres.
Inscrivez vous !
Organisez près de chez vous !
🗓 Samedi 6 juin 📷 20 h 00 📷Paris
https://www.helloasso.com/associations/reinfo-liberte/evenements/cinema-diffusion-du-film-an-inconvenient-study-du-realisateur-del-bigtree
🗓Dimanche 7 juin 📷 11 h 00 📷Paris
https://urls.fr/sZbzww
🗓 Dimanche 7 juin 📷 16 h 30 📷 Lyon
https://billetweb.fr/cine-debat-une-etude-qui-derange
Pour organiser une séance près de chez vous:
meha-france@proton.me



🎙️ **Conseil Scientifique Indépendant (CSI)** – Corruption systémique dans les sciences biomédicales : le secret de Polichinelle Avec le Dr Louis Fouché (médecin anesthésiste-réanimateur, éthique et anthropologie de la santé), le sociologue Laurent Mucchielli (directeur de recherche CNRS) et le Pr Philippe Brouqui (maladies infectieuses et tropicales). Dans cet épisode, Laurent Mucchielli décortique la **corruption systémique** qui gangrène les sciences biomédicales : conflits d’intérêts massifs avec l’industrie pharmaceutique, fraudes dans les essais cliniques, biais dans les grandes revues (LancetGate, masques, hydroxychloroquine), modélisations catastrophistes erronées, rôle des agences sanitaires (OMS, EMA, HAS) et des partenariats public-privé. Un « secret de Polichinelle » connu de longue date, amplifié pendant la crise du Covid, qui pose la question fondamentale de la confiance dans la science aujourd’hui. Philippe Brouqui et Louis Fouché réagissent et esquissent des pistes pour sortir de cette impasse : radicalité dans l’évaluation des conflits d’intérêts, retour des universités dans la production de savoir indépendant, valorisation des préprints, restauration de l’esprit critique. Un échange dense, documenté et indispensable pour comprendre comment la production du savoir scientifique a été capturée et comment reconstruire une science digne de ce nom. 📌 **Thèmes principaux :** • Corruption systémique et délinquance en col blanc dans la recherche biomédicale • Conflits d’intérêts économiques et politico-idéologiques • Fraudes scientifiques et biais méthodologiques pendant le Covid • Rôle des grandes revues et des agences sanitaires • Perte de crédit symbolique de la science et montée des algorithmes • Pistes pour une science plus indépendante et éthique 🔗 Article de Laurent Mucchielli : « Corruption systémique dans les sciences bio-médicales : le secret de Polichinelle » (disponible sur HAL) https://shs.hal.science/halshs-05566756 Abonnez-vous pour plus d’analyses indépendantes sur la santé, la science et l’éthique. #CorruptionScientifique #SciencesBiomédicales #BigPharma #ConflitsDIntérêts #Covid19 #LancetGate #ÉthiqueScientifique #LaurentMucchielli #LouisFouché #CSI #SecretDePolichinelle #SantéPublique — 💬 En commentaire : pensez-vous que la science peut encore être sauvée de cette corruption systémique ? Quelles solutions proposez-vous ? Likez, partagez et activez la cloche 🔔 pour soutenir le débat libre et contradictoire.
Dans cet épisode du Conseil Scientifique Indépendant, Louis Fouché reçoit Annette Lexa, toxicologue experte judiciaire, inscrite sur la liste Eurotox et auprès du tribunal judiciaire de Metz, avec plus de 40 ans d’expérience en toxicologie réglementaire.
Annette Lexa explique la différence fondamentale entre science académique et science réglementaire, et décortique avec précision les documents réglementaires européens (Risk Management Plans, guidelines OMS, AMM conditionnelle) qui ont encadré le développement des vaccins à ARN messager en 2020-2021.
Elle révèle :
L’absence totale de lignes directrices adaptées aux vaccins ARN au moment de leur mise sur le marché
Les manquements graves en phase préclinique (pharmacocinétique, toxicité, qualité, stabilité)
Les seuils d’acceptabilité d’impuretés et de risques très différents entre médicaments et substances chimiques
Le choix contestable du principe actif (ARN seul vs nanolipides + ARN)
La précipitation, l’impréparation et les pressions qui ont conduit à une autorisation conditionnelle malgré des données incomplètes
Un exposé technique mais clair sur la science réglementaire, souvent ignorée, qui montre que toutes les alertes étaient déjà dans les documents officiels dès 2020-2021.
Avec la participation d’Eric Ménat.
Liens utiles :
→ Fil X (Twitter) d’Annette Lexa : recherche « Annette Lexa »
→ Conseil Scientifique Indépendant
Si vous voulez comprendre comment la réglementation a été contournée et pourquoi tant de questions posées dès le début se sont révélées justifiées, cet entretien est essentiel.
Partagez, likez et commentez !
#ScienceRéglementaire #VaccinsARN #AnnetteLexa #LouisFouché #Toxicologie #AMMConditionnelle #ConseilScientifiqueIndépendant
![]()
Partenaires
![]()
![]()
![]()
Suivez-nous sur les réseaux !
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()